2019年12月以来,新型冠状肺炎疫情蔓延全球,给世界各国人民健康带来巨大威胁,并严重阻碍了经济发展。新冠疫情之所以如此复杂,一个重要原因在于病毒会变异,变异后的变种病毒特性难以捉摸。到目前为止,我们熟知的变种已经有阿尔法(Alpha)、贝塔(Beta)、 德尔塔(Delta)、奥米克戎(Omicron)等。事实上,这些仅是“闯出了名堂”的变种,那些没形成气候的变种更是多达几千种以上。科学家们对这些变种进行了归类,并为每一类取了个名字,比如德尔塔病毒叫B.1.617.2,奥米克戎病毒叫B.1.1.529等。这一命名规则称为PANGO命名法。
那么,这些被统称为“新冠”的各种病毒中,哪些传染性更强呢?传统方法多采用流行病学方法,通过溯源病例的传播途径来统计传染强度,一般称为基础再生数(R),简单地说就是一个病例会传染多少人。这种方法显然有点儿事后诸葛亮的意思,不利于对强传染性变种的早期预警。2022年5月24日 ,MIT和哈佛大学等单位在《科学》杂志上联合发表了一项研究成果,利用病毒本身的基因序列来预测它的传染性。
科学家们采用一种称为“贝叶斯逻辑斯蒂回归(Bayesian Logistic Regression)”的机器学习模型,输入为基因序列,输出为某一时间-空间点上不同新冠病毒变种的相对传染能力(Relative fitness) 。我们简称这一模型为M-H模型。为构造M-H模型,科学家们首先从GISAID数据库中得到6,466,300条基因序列,涵盖1560个地区,32个时间段(2个星期一个时间段)。对这些基因数据以PANGO命名为基础分成3000个类型。模型以基础序列的变异情况作为输入(图中黑白矩阵),同时将地区和时段作为条件变量(图中三维魔方),预测3000类病毒的相对传染能力(图中彩色曲线)。模型训练完成以后,即可以对病毒的传染能力和传播趋势做出很多重要预测。
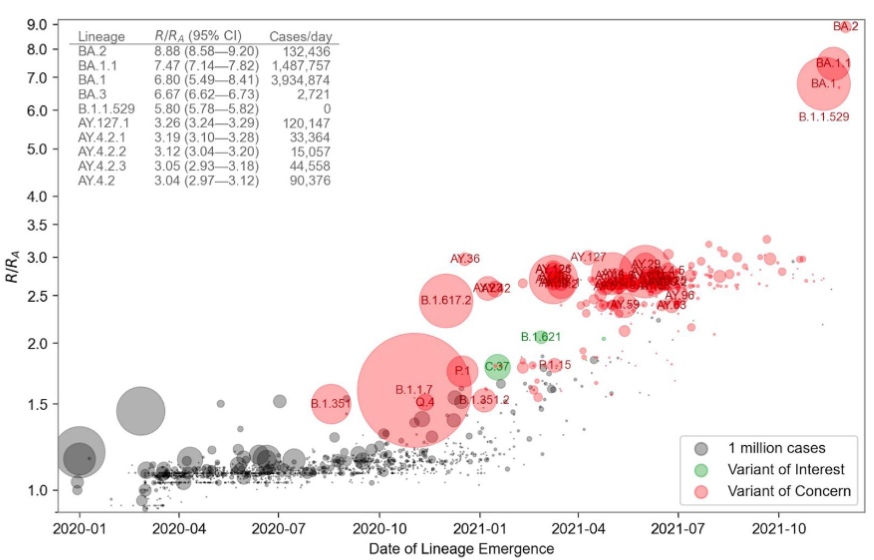
研究者利用M-H模型对各个病毒变种的传播能力进行了研究,结果如下图所示。图中横轴为时间,纵轴为以相对基础再生数(R/RA,其中RA是武汉变种的基础再生数)代表的传染能力。图中每个圈代表一个变种,其横轴的位置是其出现的时间,纵轴的位置是预测得到的传染能力,而圈的大小代表病例。
首先看到,越是后来出现的变种,其传染性越强。2021年底出现的奥米克绒变种BA.1.1,其传染性(基础再生数R)已经是武汉变种的8倍,而其后出现的BA.2变种传染力进一步加强。越新的变种传染力越强,这是疫情到目前为止依然复杂的原因。
另外,研究者发现上述AI模型确实准确地预测出了几次较大规模的传播,如2020年底由Alpha(B.1.1.7)和Delta(B.1.617.2)变种引起的爆发。图中红色圆圈代表产生较大影响的变种。
利用M-H模型,可以定位新冠病毒基因序列中对传染性影响最大的基因点。这是因为在模型设计时,科学家们为每个变异点都设计了一个可学习的显著值,学习结束后就可以通过这些显著值发现那些显著值最大的基因了。下图是科学家们发现的一些显著基因。有了这一信息,如果再来一个新变种,通过观察这些显著基因是否发生了改变,就可以预测这一变种的传染能力。如果发现有好多显著基因发生了改变,那就要加倍小心,可能又一波疫情要来了。

参考文献:
[1]https://www.science.org/doi/epdf/10.1126/science.abm1208
By:清华大学 王东